-
E-MAIL
uscert@naver.com

-
E-MAIL
uscert@naver.com
-
TEL
02-529-8005

-
TEL
02-6226-9776


 KOR
KOR  ENG
ENG  JPN
JPN  CHN
CHN 

글로벌 RA 인허가
 HOME > 글로벌 RA 인허가 > 미국
HOME > 글로벌 RA 인허가 > 미국
미국
미국(FDA)에서 정의한 의료기기란
미국에서 의료기기는 FD&C법의 Section 201(h)에서 다음과 같이 정의되어 있습니다.
• [Section 201] (h)
‘의료기기’란 기계, 기구, 도구, 장치, 삽입물, 체외 시약 또는 기타 유사하거나 관련된 물품으로 다음과 같은 모든 부속품 또는 액세서리를 포함한다.
- 1) 공식국가 처방서, 또는 미국 약전, 또는 그 모두에 관한 변경 문서에 기록된 것
- 2) 사용 목적이 인간 또는 기타 동물의 질병, 기타 상태의 진단, 치료, 경감 또는 예방인 것
- 3) 인체 또는 동물의 체내 구조 또는 기능에 영향을 미치는 것으로 체내 화학작용을 통해 주요 목적을 이루지 않고 그 목적 달성을 위해
신진 대사 작용에 영향을 받지 않는 것
• FDA 의료기기 등급 체계 및 인증 진행 방법
미국에서 체외진단용 의료기기의 등급 체계는 의료기기(MDD)와 마찬가지로 안전성 및 유효성 등을 고려한 위험도 수준에 따라 Class I, II, III로 분류합니다.
| 등급 | 의료기기 범위 | 적용규제 | 인증취득 진행방법 |
|---|---|---|---|
| Class I | 보편적으로 다른 장비들보다 설계에 있어서 단순하여 사용자에 대한 위험성이 낮은 저/중등도 위험 의료기기 (전체 의료기기의 약 30%) |
1) 일반규제 2) GMP (해당 시) |
주로 시판 전 등록: Registration |
| Class II | 일반규제(General control)만으로 안전성과 효능을 확인하기에 불충분한 중등도~고위험 의료기기 (전체 의료기기의 약 60-70%) | 1) 일반 및 특별규제 2) GMP (해당 시) |
주로 시판 전 신고: 510(k) |
| Class III | 삽입용/ 이식용 의료기기, 생명 유지용 의료기기와 같이 일반규제와 특별규제로도 안전성과 효능을 확인하기에 불충분한 고위험 의료기기 (전체 의료기기의 10% 미만) | 1) 일반 및 특별규제 2) GMP 3) 임상시험 (해당 시) |
주로 시판 전 승인: PMA |
미국(FDA)에서 요구하는 시설등록(Establishment Registration)
우리나라의 ‘업허가’ 와 비슷한 성격을 가지는 규제 절차이며, 미국 내에 판매 또는 임대를 목적으로 의료기기를 유통하기 위해서 제조원 및 수입원은 [21CFR 807 Subpart B/C]에 해당하는 절차에 따라 의료기기의 생산 및 유통에 관련된 시설을 FDA에 등록해야 합니다.
- - 제조업자 등록에 필요한 정보 : 제조원의 상호명, 주소, 연락처 / 미국 내 US Agent의 상호명, 주소, 연락처 / 미국 내 수입자의 상호명, 주소, 연락처
- - 제조업자 등록을 마친 후, 제품 등록을 하기 전에 해당 제품을 수입하려는 수입업자도 수입업자등록(Initial importer Registration)을 통하여 별도로 등록되어 있어야 합니다.
• FDA Establishment Registration(시설등록) 대상
국내/외 의료기기 제조업체(Manufacturer)
의료기기 포장업체(Re-packer & Re-labeler)
• FDA Establishment Registration(시설등록) 확인사항
의료기기 제조원 시설등록은 시설 및 제품에 대한 승인을 의미하는 것은 아니므로 시설 등록을 완료했다고 하더라도 시설 및 제품에 대한 승인은 따로 진행해야 합니다.
510(k) 또는 PMA를 진행하기 전에 공장 및 시설 등록이 되어 있어야 하지는 않지만, 최소한 기기 판매 30일 전에는 관련 시설들이 등록되어야 합니다.
• FDA Class I Device Registration
Class I 제품은 제품의 위험도가 낮기 때문에 허가가 아닌 등록의 개념입니다. 제조업자 및 수입업자 등록, 제품 등록이 필요하며 FDA에서 규정하는 일반 규제에 따라서 등록하게 됩니다.
• FDA Class I Device Registration 대상
국내/외 Class I 의료기기 제조업체(Manufacturer)
Class I 의료기기 포장업체(Re-packer & Re-labeler)
Class I 의료기기 수입업체(Importer)
• FDA Class I Device Registration 등록 절차

• FDA Class II(510(k)) Device Registration 대상
Class II device의 Submission은 ‘시판 전 신고(Pre-market Notification)’ 절차를 거쳐야 하며 통상적으로 510k라고 부릅니다.
Class II 제품들은 새롭게 신고하려는 제품의 본질적 동등성(Substantial Equivalence, SE)을 기존 허가 제품들(Predicate device)과 비교했을 때,
사용 목적과 성능이 동일하며 관련된 기술이 안전성 및 유효성에 문제를 일으키지 않는다는 것이 확인된 제품에 대하여 510k 제품 심사 절차가 적용 가능합니다.
허가를 받고 나면 GMP 대상이 되며 GMP는 허가 후 2-3년 주기로 무작위로 선정되어 진행됩니다.
※ 510(k)허가는 미국으로의 수출을 희망하는 기업에는 필수이며, 이미 FDA 허가를 받은 유사 제품과의 동등성 (SE)이 510(k) 허가에 중요한 판단 요인이 됩니다.
• FDA Class II510(k) 심사 종류
- 1) Traditional : Predicate device와 사용 목적이 동일하고 본질적 동등성에 영향을 주지 않는 선에서 신청자가 제출한 적이 없는 ‘새로운 의료기기’ 가 해당합니다. 분류상 510(k)에 해당하는 경우, 가장 본질적으로 동등한 제품(SE)과 비교하여 진행합니다.
- 2) Special : Predicate device와 사용 목적이 동일하고 본질적 동등성에 영향을 주지 않는 선에서 기기에 변경이 있는 경우 적용 가능하며, 변경사항이 중대한 안전이나 유효성에 문제가 없는 ‘설계’ 또는 ‘부품 변경’ 인 경우에만 해당됩니다. 기존 Traditional 510(k)와 동일한 포맷으로 진행하나 변경된 부분을 중점적으로 평가 및 심사 작업 등이 진행되기 때문에 비교적 신속하게 진행됩니다.
- 3) Abbreviated : 안내지침서 및 특별 규제 요건이 존재하는 경우 혹은 FDA에 의하여 관련 인정기준에 적합하다는 것을 인정받은 경우 진행 가능한 절차이며 평가 및 심사 작업 등이 신속하게 진행됩니다
• FDA Establishment Registration(시설등록) 확인사항
제품 등급 및 제품 코드, SE 검색 및 확인
시설등록 대행
제품 등록 대행
제품 라벨 컨설팅
US Agent 역할 수행
연간 등록 업무 대행
• FDA Class II(510(k)) Device Registration 대상
국내/외 Class II510(k)의료기기 제조업체(Manufacturer)
Class II510(k)의료기기 포장업체(Re-packer & Re-labeler)
• FDA Class I Device Registration 등록 절차
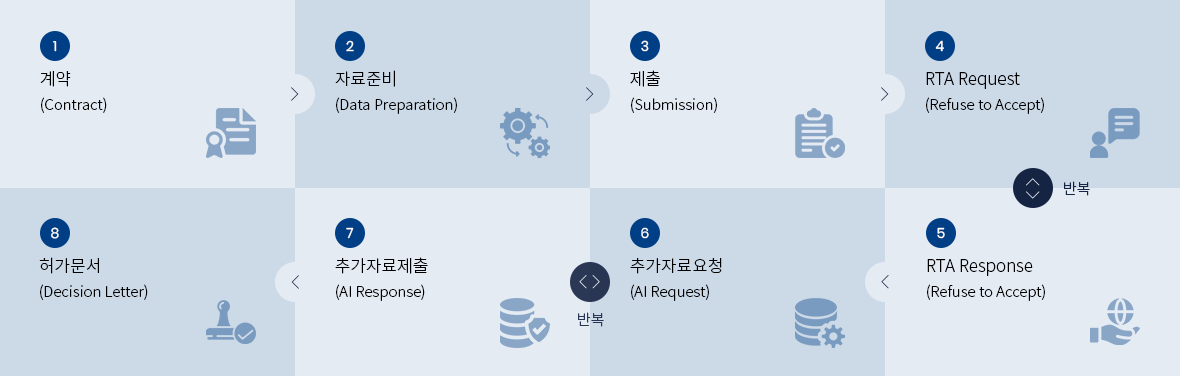
• FDA Class III(PMA) Device Registration
의료기기 중에서 가장 위험도가 높게 평가되는 의료기기로서, ‘시판 전 승인’ 절차를 거쳐야 하며 통상적으로 PMA submission이라고 부릅니다.

U.S인증원은 미국(FDA) 의료기기 제조 또는 수출 인·허가와 관련하여 고객의 요구와 법적 요구사항을 충족할 수 있도록
관련 기관과의 협업을 통하여 맞춤형 기술지원이 가능합니다.
INFO
U.S Cert Co. Ltd
Business License Number
456-81-00920
Business License Number
456-81-00920
ADDRESS
16. Disital-ro 32ga-gil. Guro-gu
Seoul, Republic of Korea (08393)
Gregory K
Seoul, Republic of Korea (08393)
Gregory K
CONTACT
T. 82-2-529-8005
E. uscert@naver.com
E. uscert@naver.com




COPYRIGHT USCERT ALL RIGHTS RESERVED.