-
E-MAIL
uscert@naver.com

-
E-MAIL
uscert@naver.com
-
TEL
02-529-8005

-
TEL
02-6226-9776

 韩语
韩语  英语
英语  日语
日语  中文
中文 

全球 RA 许可证
 HOME > 全球 RA 许可证 > 韓国
HOME > 全球 RA 许可证 > 韓国
韓国
Korea (MFDS) Medical Device Act
In Korea, there is the Medical Device Act, which stipulates the basis of laws such as the definition and permission of medical devices, and is subdivided into enforcement ordinances and enforcement rules, and is composed as follows.
Medical device as defined in Korea (Medical Device Act Article 2 Paragraph 1 - Paragraph 3 Definition of Medical Device)
Appliances, machines, devices, materials, or similar products used alone or in combination with humans or animals that fall under any of the following
Products used for the purpose of diagnosis, treatment, alleviation, treatment or prevention of disease
Products used for the purpose of diagnosis, treatment, alleviation or correction of an injury or disability
Products used for the purpose of inspection, replacement or transformation of structure or function;
Products used for the purpose of controlling pregnancy
(※However, drugs and quasi-drugs according to the Pharmaceutical Affairs Act and prosthetics and orthosis are excluded from among the assistive devices for the disabled according to Article 65 of the 「Welfare Act for the Disabled」)
• Classification of medical devices (Regulations on medical device items and grades by item (Ministry of Food and Drug Safety Notice No. 2018-15))
Medical devices are classified into four classes according to the intended use and the degree of potential harm to the human body when used
(Criteria for judging potential risk: period of contact with the human body / degree of invasion / whether drugs and energy are delivered to the patient / whether there is a biological effect)
Class 1 - Medical device with little potential risk
Class 2- Low Potentially Hazardous Medical Devices
Class 3 - Medical devices with potential hazards of moderate severity
Class 4 - High-risk medical devices
The licensing system operated by Korea (MFDS)
• Pre-marketing
As manufacturers, the subjects of pre-market regulation are clinical trials that evaluate the safety and effectiveness of products for market entry, product notification, certification and approval, and GMP conformity certification, which is a manufacturing system manager, and are managed separately.
• Post-marketing
Post-marketing regulation is followed up through advertisement pre-deliberation system, side effects/safety reporting, and re-evaluation/re-examination.
Medical device licensing procedure as defined by Korea (MFDS)
• 1st grade
Item declaration (immediately processed by the Medical Device Information Technology Support Center)
- 1) Registration of item notification application [Medical Device Electronic Complaint Counter] → Accept the report immediately upon completion of registration
- 2) Contents of technical documentation: name (product name, item name, model name) / classification number (grade) / shape and structure / purpose of use / How to use / Precautions for use / Manufacturer (in case of import or consignment of manufacturing process) / Remarks
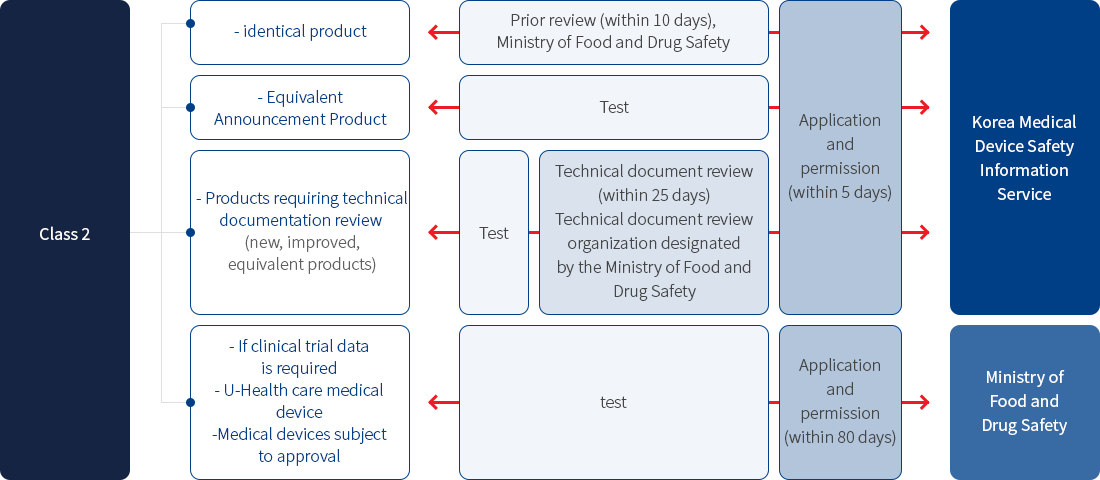
• 2nd grade
Item certification (depending on whether clinical testing is required, conducted by the Medical Device Safety Information Service or the Ministry of Food and Drug Safety)
- 1) Item classification criteria
- 2) Classification criteria for equivalent products: purpose of use, principle of action, raw materials, performance, test standards, and methods of use (excluding judgment criteria for raw materials in the electrical field)
- 3) New product: Medical device that is not equivalent to the medical device that has already been approved for use, principle of action, or raw materials (limited to medical supplies)
- 4) Improved product: A medical device that has the same purpose, principle of action, and raw material (limited to medical supplies) as that of a medical device that has already been approved, but is not identical in performance, test standards, and method of use.
- 5) Equivalent products: Medical devices that have the same purpose, principle of action, raw materials (limited to medical supplies), performance, test standards, and methods of use, etc., to medical devices that have already been approved
-
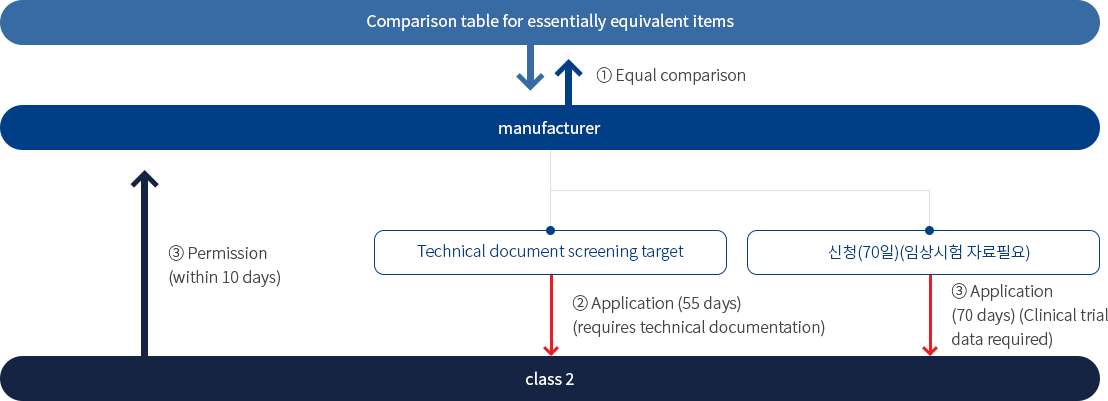
• Grades 3 and 4
Approval of items (inclusive review and proceeding by the Ministry of Food and Drug Safety Headquarters)
- 1) Level 2 and flow are the same. The period for reviewing technical documents is 55 days, and 65 days for a batch application for permission, there is a difference in period
-
GMP in Korea (MFDS)
Medical device GMP specifies the requirements of the quality management system applied to the design, development, production, installation and service of medical devices. Detailed GMP requirements can be found in 「Notice on Medical Device Manufacturing and Quality Control Standards」.
Applicable to all medical device manufacturers and importers, the type of examination is divided into document examination and on-site examination.
After the initial review, you must undergo a periodic renewal review every three years. If additional items other than previously certified items occur among the item groups classified by GMP, or if the location is changed, additional on-site inspection may be required.

美国 CERT 可以通过与相关组织合作,提供专业的技术支持,以满足客户在韩国制造和销售体外诊断医疗器械的需求和法律要求。此外,我有信心通过成功获得国内体外诊断医疗器械许可证,重生为一家有前途的公司
情报
U.S Cert Co. Ltd
营业执照号
456-81-00920
营业执照号
456-81-00920
地址
16. Disital-ro 32ga-gil. Guro-gu
Seoul, Republic of Korea (08393)
Gregory K
Seoul, Republic of Korea (08393)
Gregory K
联系方式
T. 82-2-529-8005
E. uscert@naver.com
E. uscert@naver.com



RESERVED. (版权所有USCERT保留所有权利。)