-
E-MAIL
uscert@naver.com

-
E-MAIL
uscert@naver.com
-
TEL
02-529-8005

-
TEL
02-6226-9776

 韩语
韩语  英语
英语  日语
日语  中文
中文 

全球 RA 许可证
 HOME > 全球 RA 许可证 > 美国
HOME > 全球 RA 许可证 > 美国
美国
What is a medical device as defined by the United States (FDA)
In the United States, a medical device is defined in Section 201(h) of the FD&C Act as:
• [Section 201] (h)
“Medical device” is a machine, instrument, instrument, device, insert, in vitro reagent or other similar or related article, including all accessories or accessories as follows:
- 1) As recorded in the official national prescription, or in the United States Pharmacopoeia, or a change document to both.
- 2) The purpose of use is the diagnosis, treatment, alleviation or prevention of diseases or other conditions in humans or other animals
- 3) A substance that affects the structure or function of the human body or animal, does not achieve its main purpose through chemical reactions in the body, and is not affected by metabolism in order to achieve that purpose
• FDA Medical Device Rating System and Certification Process
In the United States, the classification system for in vitro diagnostic medical devices is classified into Class I, II, and III according to the level of risk in consideration of safety and effectiveness, just like medical devices (MDD).
| class | Medical Device Scope | Applicable regulations | How to obtain certification |
|---|---|---|---|
| Class I | Low/moderate risk medical devices with low risk to users due to their simplicity in design compared to other devices in general (about 30% of all medical devices) | 1) General regulations 2) GMP (if applicable) |
Registration Mainly pre-market registration: Registration |
| Class II | Moderate to high-risk medical devices that are insufficient to confirm safety and efficacy through general control alone (About 60-70% of all medical devices) | 1) General and special regulations 2) GMP (if applicable) |
Primarily premarket notification: 510(k) |
| Class III | High-risk medical devices that are insufficient to confirm safety and efficacy even with general and special regulations, such as implantable/implantable medical devices and life-sustaining medical devices (less than 10% of all medical devices) | 1) General and special regulations 2) GMP 3) Clinical trial (if applicable) |
Primarily premarket approval: PMA |
Establishment registration required by the US (FDA)
It is a regulatory procedure similar to Korea's 'business license', and in order to distribute medical devices for the purpose of sale or rental in the United States, manufacturers and importers must Facilities involved in production and distribution must be registered with the FDA.
- - - Information required for manufacturer registration: Manufacturer's name, address, contact information / US Agent's trade name, address, contact information / Importer's name, address, and contact information in the United States
- - After completing the manufacturer registration, before registering the product, the importer who wants to import the product must also be separately registered through Initial importer Registration.
• For FDA Establishment Registration
Domestic/foreign medical device manufacturers (Manufacturer)
Medical device packaging company (Re-packer & Re-labeler)
• FDA Establishment Registration Checklist
Medical device manufacturer facility registration does not mean approval of facilities and products, so even if facility registration is completed, approval for facilities and products must be carried out separately.
Plants and facilities are not required to be registered prior to proceeding with a 510(k) or PMA, but must be registered at least 30 days prior to device sale.
• FDA Class I Device Registration
Class I products are a concept of registration, not permission, because the product has a low risk. Manufacturer and importer registration, product registration is required, and registration is performed according to the general regulations stipulated by the FDA.
• Subject to FDA Class I Device Registration
Domestic/foreign Class I Medical Device Manufacturer (Manufacturer)
Class I medical device packaging company (Re-packer & Re-labeler)
Class I medical device importer
• FDA Class I Device Registration Registration Process

• Subject to FDA Class II (510(k)) Device Registration
Class II products have the same purpose and performance when comparing the Substantial Equivalence (SE) of the new product to be declared with the existing predicate device (Predicate device). The 510k product screening process is applicable for identified products. After obtaining permission, it is subject to GMP, and GMP is randomly selected every 2-3 years after approval.
※ A 510(k) permit is essential for companies wishing to export to the United States, and the equivalence (SE) with a similar product that has already been approved by the FDA is an important factor in determining the 510(k) permit.
• FDA Class II510(k) Review Type
- 1) Traditional : A ‘new medical device’ that has not been submitted by the applicant as long as the purpose of use is the same as that of the predicate device and does not affect the essential equivalence. If it is classified as a 510(k), it proceeds against its most essentially equivalent product (SE).
- 2) Special : It is applicable if there is a change in the device as long as the purpose of use is the same as the predicate device and does not affect the essential equivalence, and the change is a 'design' or 'part change' without significant safety or effectiveness problems. Only in case. It proceeds in the same format as the existing Traditional 510(k), but it proceeds relatively quickly because evaluation and examination are conducted focusing on the changed parts.
- 3) Abbreviated : This is a process that can be proceeded when there are guidelines and special regulatory requirements, or when it is recognized that the relevant accreditation standards are met by the FDA, and evaluation and review work is carried out quickly.
• FDA Establishment Registration Checklist
Search and verify product grades and product codes, SEs
Facility registration agency
Product registration agency
Product label consulting
US Agent role
Annual registration service
• Subject to FDA Class II (510(k)) Device Registration
Domestic/foreign Class II510(k) medical device manufacturer (Manufacturer)
Class II510(k) medical device packaging company (Re-packer & Re-labeler)
• FDA Class II (510(k)) Device Registration Process
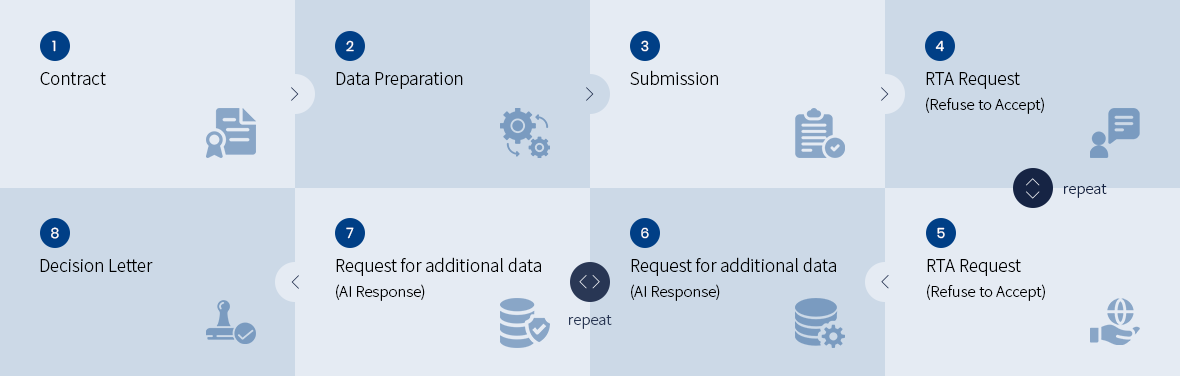
• FDA Class III (PMA) Device Registration
As a medical device with the highest risk among medical devices, it has to go through a ‘premarket approval’ procedure and is usually called a PMA submission.

美国 CERT 能够通过与相关组织的合作,提供定制的技术支持,以满足客户在出口许可证和医疗器械对华许可证 (CFDA (NMPA)) 方面的需求和法律要求
情报
U.S Cert Co. Ltd
营业执照号
456-81-00920
营业执照号
456-81-00920
地址
16. Disital-ro 32ga-gil. Guro-gu
Seoul, Republic of Korea (08393)
Gregory K
Seoul, Republic of Korea (08393)
Gregory K
联系方式
T. 82-2-529-8005
E. uscert@naver.com
E. uscert@naver.com



RESERVED. (版权所有USCERT保留所有权利。)